Why RNA viruses dominate pandemic history — by the numbers
Every major pandemic of the last century — the 1918 Spanish flu, the 2009 H1N1 swine flu, SARS, MERS, Ebola, Zika, HIV, and COVID-19 — was caused by an RNA virus. This is not coincidence. RNA viruses mutate at rates roughly 10,000 times faster than DNA viruses: while a DNA virus introduces around one error per 100 million nucleotides copied, an RNA virus makes one mistake every 10,000 to 1,000,000 nucleotides. That single molecular difference — the absence of a genetic proofreading enzyme — generates a cloud of variants with every infection, fuelling the rapid evolution, immune escape, and cross-species jumps that define pandemic pathogens. This article unpacks the molecular biology behind that number, and explains why it matters for surveillance, vaccines, and antivirals.
Section 1 — The numbers don’t lie: viral mutation rates
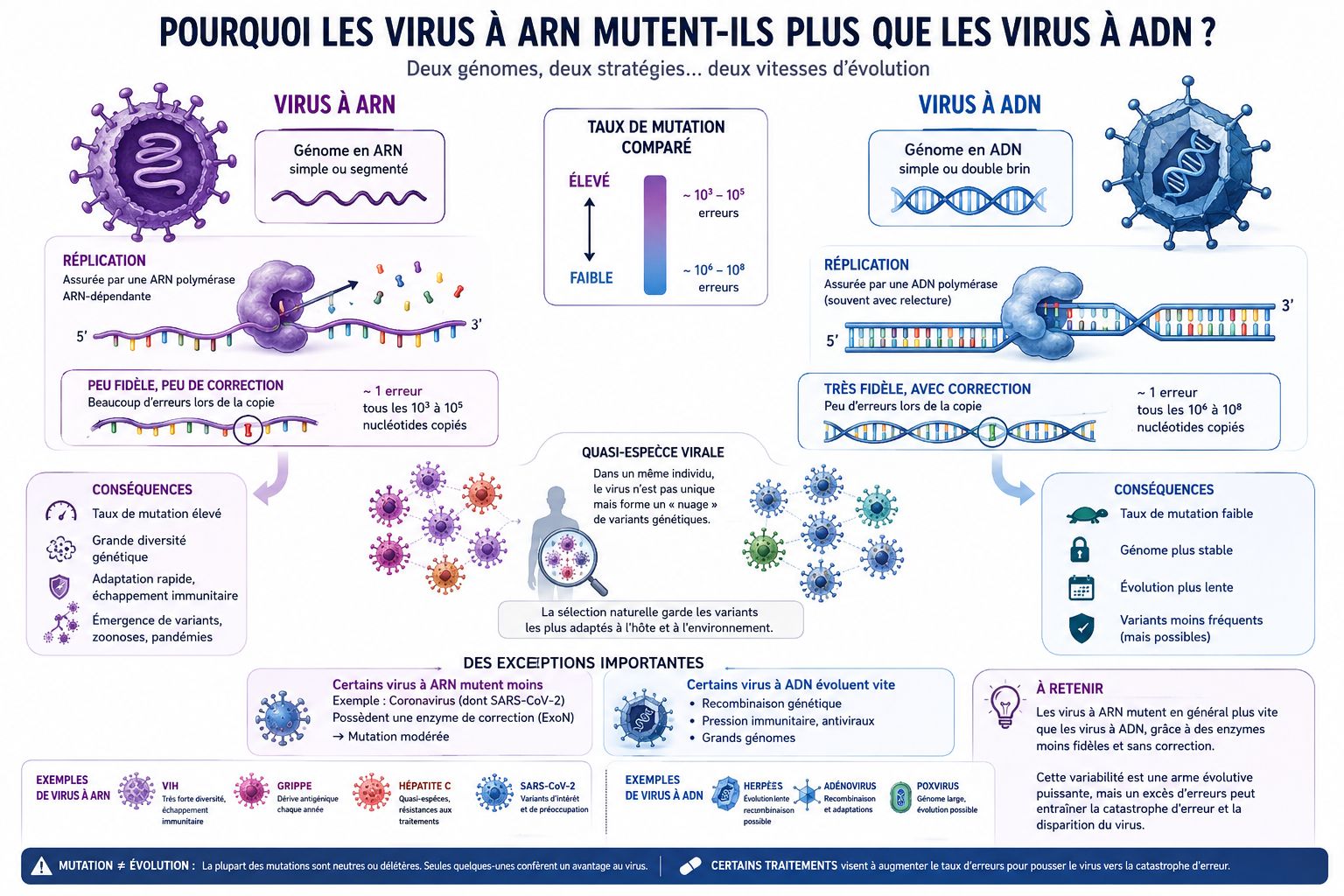
To understand why RNA viruses are so disproportionately represented among pandemic pathogens, start with the most fundamental measurement in virology: the mutation rate — the number of copying errors introduced per nucleotide (the letter in the genetic code) per replication cycle. In 2010, Sanjuán and colleagues published the landmark meta-analysis « Viral Mutation Rates » in the Journal of Virology, pooling data from more than 40 studies spanning 23 viruses. Their conclusion was unambiguous: RNA viruses mutate at rates between 10⁻⁶ and 10⁻⁴ substitutions per nucleotide per cell infection (with a geometric mean of 2.2 × 10⁻⁵). DNA viruses, by contrast, cluster between 10⁻⁸ and 10⁻⁶ — and crucially, the two distributions show no overlap whatsoever.
To translate those exponents into everyday language: if you copied the human genome at the error rate of influenza A (2.3 × 10⁻⁵ per nucleotide, per the same Sanjuán dataset), you would introduce roughly 70,000 mutations every time a single cell divides. DNA repair mechanisms in human cells keep our own mutation rate many orders of magnitude lower. Herpes simplex virus type 1 (HSV-1), a DNA virus, clocks in at 5.9 × 10⁻⁸ — essentially the same tier as cellular DNA replication. The gap between influenza and HSV-1 is approximately 400-fold; between hepatitis C virus (HCV, 1.2 × 10⁻⁴) and HSV-1, it reaches five orders of magnitude.
A comprehensive 2021 review by Domingo and colleagues in Viruses — led by Esteban Domingo, one of the founders of quasi-species theory — confirmed and extended these figures. RNA viruses produce between 10⁻³ and 10⁻⁶ copying errors per nucleotide incorporated, « one or more orders of magnitude above the replication of cellular DNA. » For SARS-CoV-2 specifically, in vivo evolution has been measured at (1.2 ± 0.5) × 10⁻³ substitutions per site per year — a rate that, during the COVID-19 pandemic, generated the Alpha, Delta, and Omicron variants within roughly 18 months of the virus’s emergence in the human population. Meanwhile, a 2012 analysis by Sanjuán in PLOS Pathogens, drawing on 37 mutation rate estimates and 223 evolutionary rate estimates across 84 viruses, confirmed that single-stranded RNA viruses and retroviruses evolve fastest, double-stranded DNA viruses slowest, with a strong correlation between mutation rate and evolutionary speed (r = 0.946, p = 0.004).
The practical consequence of this velocity is straightforward: a population of RNA viruses replicating inside a single patient generates an astronomical diversity of variants within days. Some of those variants will evade antibody neutralisation; others will replicate more efficiently in a new host species; others still will resist antiviral drugs. High mutation rates are, in evolutionary terms, the engine of RNA viral adaptability — and adaptability is precisely what makes a pandemic pathogen.
Section 2 — Proofreading: the DNA virus advantage
Why do DNA viruses mutate so much more slowly? The answer lies in a molecular editing mechanism called proofreading. Think of it as having a meticulous copy-editor review every page of a manuscript the moment it is typed: every mis-inserted letter is caught and corrected before the page is filed. DNA polymerases — the enzymes that replicate DNA — carry a built-in proofreading domain called the 3’→5′ exonuclease. This domain reads the newly synthesised strand in the reverse direction as replication proceeds, detects mismatched base pairs, excises the error, and replaces it with the correct nucleotide. The net result is a per-base error rate reduced by roughly 100-fold compared with the raw polymerisation error rate.
RNA-dependent RNA polymerases (RdRps) — the replication engines of RNA viruses — almost universally lack this proofreading capacity. As Carrasco-Hernández and colleagues noted in their 2017 review in the ILAR Journal: « The majority of RNA viral replicases lack proofreading activity. The absence of exonuclease activity increases the point mutation rate of the genome by omitting error correction during replication. » Errors accumulate freely with each replication round, generating the vast genetic diversity that characterises RNA viral populations.
There is, however, a striking exception — and it happens to be the family that caused COVID-19. Smith and Denison, writing in PLOS Pathogens in 2013, described coronaviruses as « DNA wannabes »: unlike virtually every other RNA virus, coronaviruses encode a functional proofreading exonuclease, nsp14-ExoN (nonstructural protein 14, exoribonuclease). This enzyme, part of a multi-protein replication complex also characterised by Subissi and colleagues in PNAS in 2014, provides the correction fidelity that allows coronavirus genomes to reach extraordinary sizes — up to 32 kilobases (kb) — roughly two to three times the length of a typical RNA viral genome (HIV: ~9 kb; hepatitis C: ~9.6 kb; influenza: ~14 kb across 8 segments). Without ExoN, that large genome would accumulate lethal errors far too rapidly to be viable.
The therapeutic consequence was demonstrated experimentally: Smith, Blanc, Vignuzzi, and Denison, also in PLOS Pathogens in 2013, showed that coronaviruses engineered to lack ExoN activity become up to 300 times more sensitive to the antiviral drug ribavirin, and accumulate mutations at 15–16 times the normal rate. This explains why SARS-CoV-2, unlike influenza or hepatitis C, proved relatively resistant to classical nucleoside analogues such as ribavirin — and why nsp14-ExoN has become a priority drug target for next-generation coronavirus antivirals. The coronavirus exception, far from weakening the proofreading argument, actually confirms it: the one RNA virus family that acquired proofreading capacity is the one that evolved the largest genome and the greatest architectural complexity in the RNA viral world.
Section 3 — Quasi-species theory and the error catastrophe
High mutation rates do not simply mean « more diversity. » They impose a fundamental constraint on viral existence — and that constraint has become one of the most promising targets in antiviral pharmacology. The concept is quasi-species theory, formalised by Nobel laureate Manfred Eigen and applied to RNA viruses by Domingo.
In classical biology, a viral « strain » implies a genetically homogeneous population. Quasi-species theory rejects that model for RNA viruses. Because every replication cycle introduces new mutations, an RNA viral population is better described as a mutant swarm — a cloud of closely related but genetically distinct variants gravitating around a consensus sequence. At any given moment, an infected cell harbours not one virus but a dynamic ensemble of thousands of slightly different genomes. The fittest variants dominate the swarm; the others are present at low frequencies, ready to expand if selection pressure changes (new host cell type, immune pressure, drug treatment).
This architecture has an Achilles’ heel. Eigen, in his foundational 2002 paper in PNAS, formalised what he called the error catastrophe: if the mutation rate of an RNA virus is pushed above a critical threshold — the error threshold — the accumulation of deleterious mutations overwhelms the replication fidelity of even the fittest variants, the genetic information encoding the virus is progressively degraded, and the population is driven toward extinction. RNA viruses already operate close to this threshold. Sanjuán’s 2012 analysis quantified how precarious this balance is: a mere threefold increase in mutation rate can cause « drastic fitness losses in ssRNA viruses, » and a threefold increase would translate into a 48-fold slowdown in evolutionary speed — effectively paralysing the virus’s ability to adapt.
This vulnerability is precisely what several antiviral drugs exploit. Hadj Hassine, Ben M’hadheb, and Menéndez-Arias reviewed the full landscape of lethal mutagenesis as an antiviral strategy in Viruses in 2022. The three main agents are:
- Ribavirin — the oldest RNA antiviral mutagen, used against hepatitis C and haemorrhagic fevers; its mechanism involves incorporation into viral RNA, triggering G→A and C→U transitions that accumulate beyond tolerable levels.
- Favipiravir — approved for influenza in Japan and widely used experimentally against SARS-CoV-2 and Ebola; it mimics purine bases, inserts itself into the RNA template, and amplifies transition errors across the genome.
- Molnupiravir — the prodrug β-d-N4-hydroxycytidine, approved for COVID-19 in 2021–2022, is approximately 100 times more active against SARS-CoV-2 than ribavirin. It induces C:G→T:A and G:C→A:T transitions, pushing viral genomes past the error threshold.
The quasi-species framework also explains a subtler danger: survival of the flattest. Under high-mutation-rate conditions, the evolutionary advantage does not necessarily go to the fastest replicator but to the variant whose fitness is most robust across the widest range of mutations — the flattest fitness landscape. A virus sitting on a « flat peak » in sequence space can absorb more mutations without dying, making it intrinsically harder to push over the error catastrophe cliff. This concept, described by Carrasco-Hernández and colleagues, has direct implications for drug resistance: low-fidelity RNA viral populations can evolve drug resistance with remarkable speed, sometimes with just a single point mutation.
Section 4 — Why nearly every recent pandemic has been caused by an RNA virus
The molecular biology above has a direct epidemiological translation. Woolhouse and colleagues, in a landmark 2012 paper in the Philosophical Transactions of the Royal Society B, catalogued all 219 virus species officially recognised by the International Committee on Taxonomy of Viruses (ICTV) as capable of infecting humans, and found that more than two-thirds are zoonotic — capable of moving from animal reservoirs into human populations. Among those zoonotic viruses, RNA viruses are heavily over-represented: their faster evolution equips them to bridge the molecular incompatibility between a bat immune system and a human one, between avian ACE2 receptors and human ACE2, with far greater efficiency than their DNA counterparts.
Carrasco-Hernández and colleagues estimated in 2017 that RNA viruses account for 25 to 44% of all emerging infectious diseases, despite representing a minority of known virus species by total count. Their review put the contrast in stark terms: « RNA viruses jump species boundaries more often than DNA viruses, likely because of their differing rates of evolutionary change. »
The historical record bears this out with brutal clarity. Consider the five most consequential viral pandemics of the last century:
- Influenza A (RNA, orthomyxovirus) — responsible for the 1918 Spanish flu (~50 million deaths), 1957 Asian flu, 1968 Hong Kong flu, and the 2009 H1N1 pandemic. Influenza’s segmented genome (8 separate RNA segments) enables a process called reassortment: when two influenza strains infect the same cell, their segments can shuffle like a deck of cards, producing a novel hybrid strain that human immune systems have never encountered. As detailed by Liang in Virulence in 2023, avian influenza strains H5 and H7 already carry case fatality rates exceeding 50% in humans — a pandemic threat that only remains dormant because they have not yet acquired efficient human-to-human transmission.
- HIV-1 (RNA, retrovirus) — the causative agent of AIDS; the global pandemic has claimed over 40 million lives. HIV uses a reverse transcriptase to convert its RNA genome into DNA before integration, but this enzyme is notoriously error-prone, and a single mutation confers resistance to entire classes of non-nucleoside reverse transcriptase inhibitors (NNRTIs). Mutation rate: 2.4 × 10⁻⁵ per nucleotide per cycle (Sanjuán 2010).
- SARS-CoV-2 (RNA, coronavirus) — despite having the ExoN proofreading mechanism, SARS-CoV-2 evolved Omicron BA.1 within 24 months, accumulating over 50 mutations relative to the original Wuhan sequence, including 32 in the spike protein alone — enough to substantially evade vaccine-induced immunity.
- Ebola virus (RNA, filovirus) — with case fatality rates of 25–90%, Ebola represents one of the most acutely lethal RNA viruses; its reservoir in fruit bats exemplifies the zoonotic spillover pattern documented by Keusch, Daszak and colleagues in PNAS in 2022.
- Zika virus (RNA, flavivirus) — the 2015–2016 outbreak demonstrated that RNA viral evolution could generate a neurotropic pathogen capable of causing microcephaly, a clinical complication that had never been previously associated with arboviral infection.
Keusch, Daszak and colleagues synthesised the common thread across all major RNA outbreaks since 1967: ancestral viral origins in bats, birds, or other wild mammals; intermediate animal hosts (palm civets for SARS, camels for MERS, non-human primates for Ebola); and the intensifying human-wildlife interface — deforestation, live animal markets, industrial livestock farming — that creates the conditions for spillover. Their 2022 PNAS paper explicitly framed pandemic prevention as inseparable from a « One Health » approach integrating animal, human, and environmental surveillance.
Section 5 — DNA viruses: different strategies, different dangers
Framing RNA viruses as the « dangerous » category risks a misunderstanding that deserves direct correction: DNA viruses are not harmless. They have simply evolved a different, often slower-acting, form of danger — one built on persistence, immune evasion through latency, and in several cases, oncogenesis.
Latency is the signature strategy of the herpesviruses. Herpes simplex virus type 1 (HSV-1, a DNA virus) establishes lifelong latent infection in sensory neurons after primary oral infection; it can reactivate decades later as cold sores, encephalitis, or — in immunocompromised patients — disseminated disease. Varicella-zoster virus (VZV) causes chickenpox on primary infection, then retreats to the dorsal root ganglia, where it may reactivate as herpes zoster (shingles) — sometimes accompanied by post-herpetic neuralgia lasting years — in older or immunocompromised adults. Epstein-Barr virus (EBV) persists in B lymphocytes indefinitely; it drives infectious mononucleosis acutely, and is causally linked to several cancers including Burkitt lymphoma, Hodgkin lymphoma, and nasopharyngeal carcinoma.
Oncogenesis — the direct promotion of cancer — is a domain where DNA viruses are disproportionately represented. Vojtěchová and Tachezy, in a comprehensive 2025 review in the Journal of Virology, catalogued the mechanisms by which human DNA oncoviruses integrate their genomes into host DNA. 13% of all cancers worldwide have an infectious aetiology, and DNA oncoviruses — primarily HPV, HBV, EBV, and Merkel cell polyomavirus (MCV) — account for approximately 60% of those virus-associated cancers. Human papillomavirus (HPV) oncoproteins E6 and E7 degrade the tumour suppressor p53 and inactivate the retinoblastoma protein pRb respectively, destabilising the host genome and driving progression toward cervical, oropharyngeal, anal, and other carcinomas. Hepatitis B virus (HBV) integration disrupts tumour suppressor loci and activates proto-oncogenes, contributing to hepatocellular carcinoma — the third most common cause of cancer death globally.
The contrast is thus architectural: RNA viruses threaten through speed — rapid mutation, fast evolution, explosive pandemic spread. DNA viruses threaten through persistence — decades of latency, genome integration, and the slow accumulation of oncogenic changes. Neither is « safer. » They represent different evolutionary strategies for exploiting a host organism over time.
For healthcare professionals — clinical and public health implications
- Surveillance and genomic sequencing: The quasi-species nature of RNA viruses makes real-time whole-genome sequencing (WGS) indispensable for outbreak monitoring. A single consensus sequence misses the minority variant cloud; deep sequencing reveals low-frequency drug-resistant or immune-escape mutants before they dominate the population. The rapid detection of Omicron via GISAID sequencing networks exemplified this approach.
- Reassortment risk (influenza): Co-circulation of multiple influenza A subtypes in swine or avian hosts creates reassortment hotspots. Current H5N1 clade 2.3.4.4b outbreaks in dairy cattle (US, 2024–2025) represent precisely this risk scenario — as reviewed by Liang in Virulence — requiring enhanced One Health surveillance at the livestock-wildlife-human interface.
- Antigenic drift and vaccine updates: RNA viral evolution means vaccines can become mismatched. Influenza vaccines are reformulated annually; SARS-CoV-2 XBB.1.5 and JN.1 boosters reflected the same principle. The mRNA vaccine platform — itself inspired by RNA viral biology — offers the fastest turnaround for reformulation (6–8 weeks from sequence to clinical-grade mRNA), making it uniquely suited to outpace antigenic drift.
- Antiviral resistance: Low-fidelity RNA replication means resistance mutations emerge rapidly under drug selection pressure. HIV exemplifies this: under selective pressure, resistance to lamivudine emerges at the M184V mutation within weeks. Combination antiretroviral therapy (cART) was developed specifically to overcome single-mutation resistance by targeting multiple steps simultaneously. The same principle now guides COVID-19 antiviral strategy (protease inhibitor + RdRp inhibitor combinations).
- Lethal mutagenesis as a clinical strategy: Molnupiravir’s approval for COVID-19 in high-risk patients represents the clinical translation of error-catastrophe theory. However, concern exists around host genotoxicity — the drug’s mutagenic mechanism is not perfectly selective for viral RNA, raising questions about potential off-target effects in rapidly dividing host cells, as discussed by Hadj Hassine and colleagues. Ongoing pharmacovigilance remains warranted.
- DNA virus oncogenesis — preventable cancers: The HPV vaccine (Gardasil 9) targets the nine most oncogenic HPV genotypes and is the most successful viral cancer prevention tool in history. HBV vaccination at birth, now part of routine childhood schedules in most countries, prevents the vast majority of HBV-related hepatocellular carcinomas. Ensuring universal coverage of both vaccines is among the highest-yield oncology interventions available, as the Vojtěchová and Tachezy review underscores.
- Biosecurity and One Health: Keusch, Daszak and colleagues estimate that between 650,000 and 840,000 zoonotic viruses with pandemic potential remain uncharacterised in wildlife reservoirs. Systematic surveillance of animal–human interfaces, pandemic preparedness investment, and policies that reduce the deforestation and live-animal trade drivers of spillover are the structural interventions most likely to prevent the next RNA pandemic.
Conclusion — molecular biology as pandemic policy
RNA viruses do not cause more pandemics than DNA viruses out of malice or special virulence. They do so because of a single molecular accident of evolutionary history: the RNA-dependent RNA polymerases that replicate their genomes lack the proofreading exonuclease that DNA polymerases acquired hundreds of millions of years ago. That absence generates mutation rates 10,000 times higher, quasi-species swarms in every infected host, and an evolutionary agility that allows these viruses to cross species barriers, evade immune memory, and generate pandemic-capable strains on timescales of months rather than millennia.
The coronavirus exception — a family of RNA viruses that invented their own proofreading — only reinforces the rule: fidelity has a cost (genome size is constrained) and a benefit (reduced mutation rate, greater genome complexity). SARS-CoV-2 still evolved faster than any DNA virus, simply more slowly than influenza. The pandemic consequences were devastating regardless.
Understanding this biology is not merely academic. It explains why influenza vaccines must be reformulated every year, why HIV therapy requires combinations of drugs targeting multiple resistance barriers simultaneously, why molnupiravir works by deliberately worsening the error rate of an already error-prone polymerase, and why the mRNA vaccine platform — itself a technology borrowed from RNA viral biology — can be updated in weeks rather than months. The molecular reason behind the pandemic risk is also, encoded in it, a map of the solutions.
Key takeaways
- RNA viruses mutate at rates of 10⁻⁶ to 10⁻⁴ substitutions per nucleotide per infection cycle — roughly 10,000 times faster than DNA viruses — with no overlap between the two distributions in experimental data (Sanjuán et al., J. Virol. 2010).
- The root cause is the absence of a proofreading exonuclease in RNA-dependent RNA polymerases; DNA polymerases possess this 3’→5′ error-correction domain, reducing their effective error rate by ~100-fold.
- Coronaviruses are the notable exception: the nsp14-ExoN exonuclease provides partial proofreading fidelity, enabling the ~30 kb coronavirus genome — the largest in the RNA viral world — and explaining the partial resistance of SARS-CoV-2 to classical mutagenic antivirals.
- RNA viral populations exist as quasi-species swarms — dynamic mutant clouds — that operate close to an error catastrophe threshold; antiviral drugs such as favipiravir, ribavirin, and molnupiravir exploit this vulnerability by pushing mutation rates over that threshold toward lethal mutagenesis.
- More than two-thirds of the 219 known human virus species are zoonotic; RNA viruses account for 25–44% of all emerging infectious diseases, and every major pandemic since 1918 has been caused by an RNA virus — a pattern rooted in the molecular biology above, not coincidence.
🇫🇷 Lire cet article en français : Virus à ARN, virus à ADN : pourquoi les premiers font-ils plus de pandémies ?
Bibliography — 13 verified scientific sources
- Sanjuán R, Nebot MR, Chirico N, Mansky LM, Belshaw R. « Viral Mutation Rates. » Journal of Virology, 84(19):9733–9748, 2010. DOI: 10.1128/JVI.00694-10
- Domingo E, García-Crespo C, Lobo-Vega R, Perales C. « Mutation Rates, Mutation Frequencies, and Proofreading-Repair Activities in RNA Virus Genetics. » Viruses, 13(9):1882, 2021. DOI: 10.3390/v13091882
- Sanjuán R. « From Molecular Genetics to Phylodynamics: Evolutionary Relevance of Mutation Rates Across Viruses. » PLOS Pathogens, 8(5):e1002685, 2012. DOI: 10.1371/journal.ppat.1002685
- Smith EC, Denison MR. « Coronaviruses as DNA Wannabes: A New Model for the Regulation of RNA Virus Replication Fidelity. » PLOS Pathogens, 9(12):e1003760, 2013. DOI: 10.1371/journal.ppat.1003760
- Smith EC, Blanc H, Vignuzzi M, Denison MR. « Coronaviruses Lacking Exoribonuclease Activity Are Susceptible to Lethal Mutagenesis: Evidence for Proofreading and Potential Therapeutics. » PLOS Pathogens, 9(8):e1003565, 2013. DOI: 10.1371/journal.ppat.1003565
- Subissi L, Posthuma CC, Collet A, et al. « One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. » PNAS, 111(37):E3900–E3909, 2014. DOI: 10.1073/pnas.1323705111
- Eigen M. « Error catastrophe and antiviral strategy. » PNAS, 99(21):13374–13376, 2002. DOI: 10.1073/pnas.212514799
- Hadj Hassine I, Ben M’hadheb M, Menéndez-Arias L. « Lethal Mutagenesis of RNA Viruses and Approved Drugs with Antiviral Mutagenic Activity. » Viruses, 14(4):841, 2022. DOI: 10.3390/v14040841
- Woolhouse MEJ, Scott F, Hudson Z, Howey R, Chase-Topping M. « Human viruses: discovery and emergence. » Philosophical Transactions of the Royal Society B, 367(1604):2864–2871, 2012. DOI: 10.1098/rstb.2011.0354
- Keusch GT, Amuasi J, Anderson DE, Daszak P, et al. « Pandemic origins and a One Health approach to preparedness and prevention: Solutions based on SARS-CoV-2 and other RNA viruses. » PNAS, 119(42):e2202871119, 2022. DOI: 10.1073/pnas.2202871119
- Carrasco-Hernández R, Jácome R, López Vidal Y, Ponce de León S. « Are RNA Viruses Candidate Agents for the Next Global Pandemic? A Review. » ILAR Journal, 58(3):343–358, 2017. DOI: 10.1093/ilar/ilx026
- Liang Y. « Pathogenicity and virulence of influenza. » Virulence, 14(1):2223057, 2023. DOI: 10.1080/21505594.2023.2223057
- Vojtěchová Z, Tachezy R. « Genome integration of human DNA oncoviruses. » Journal of Virology, 99:e00562-25, 2025. DOI: 10.1128/jvi.00562-25
Article based on a review of 13 verified scientific sources (PubMed/PMC).
NutriCellScience, Mark DOWN — EN edition

Répondre à Virus à ARN, virus à ADN : pourquoi les premiers font-ils plus de pandémies ? – Nutricellscience Annuler la réponse.